Cell type annotation from marker genes
In single-cell, we have no prior information of which cell type each cell belongs. To assign cell type labels, we first project all cells in a shared embedded space, then we find communities of cells that show a similar transcription profile and finally we check what cell type specific markers are expressed. If more than one marker gene is available, statistical methods can be used to test if a set of markers is enriched in a given cell population.
In this notebook we showcase how to use decoupler for cell type annotation with the 3k PBMCs 10X data-set. The data consists of 3k PBMCs from a Healthy Donor and is freely available from 10x Genomics here from this webpage
Note
This tutorial assumes that you already know the basics of decoupler. Else, check out the Usage tutorial first.
Loading packages
First, we need to load the relevant packages, scanpy to handle scRNA-seq data and decoupler to use statistical methods.
[1]:
import scanpy as sc
import decoupler as dc
import numpy as np
# Plotting options, change to your liking
sc.settings.set_figure_params(dpi=200, frameon=False)
sc.set_figure_params(dpi=200)
sc.set_figure_params(figsize=(4, 4))
Single-cell processing
Loading the data-set
We can download the data easily using scanpy:
[2]:
adata = sc.datasets.pbmc3k()
adata
[2]:
AnnData object with n_obs × n_vars = 2700 × 32738
var: 'gene_ids'
QC, projection and clustering
Here we follow the standard pre-processing steps as described in the scanpy vignette. These steps carry out the selection and filtration of cells based on quality control metrics and the data normalization.
Note
This is just an example, these steps should change depending on the data.
[3]:
# Basic filtering
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# Annotate the group of mitochondrial genes as 'mt'
adata.var['mt'] = adata.var_names.str.startswith('MT-')
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# Filter cells following standard QC criteria.
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
# Normalize the data
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
adata.layers['log_norm'] = adata.X.copy()
Then we group cells based on the similarity of their transcription profiles. To visualize the communities we perform UMAP reduction.
[4]:
# Identify the highly variable genes
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# Regress and scale the data
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
# Generate PCA features
sc.tl.pca(adata, svd_solver='arpack')
# Restore X to be norm counts
dc.swap_layer(adata, 'log_norm', X_layer_key=None, inplace=True)
# Compute distances in the PCA space, and find cell neighbors
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=40)
# Generate UMAP features
sc.tl.umap(adata)
# Run leiden clustering algorithm
sc.tl.leiden(adata)
# Visualize
sc.pl.umap(adata, color='leiden', title='RNA UMAP',
frameon=False, legend_fontweight='normal', legend_fontsize=15)
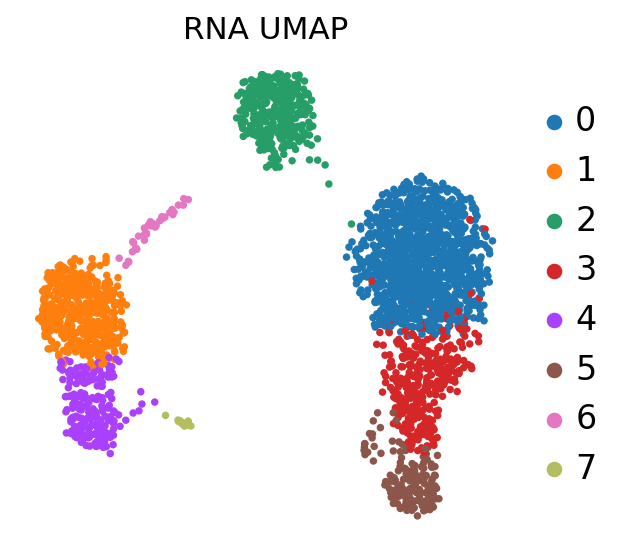
At this stage, we have identified communities of cells that show a similar transcriptomic profile, and we would like to know to which cell type they probably belong.
Marker genes
To annotate single cell clusters, we can use cell type specific marker genes. These are genes that are mainly expressed exclusively by a specific cell type, making them useful to distinguish heterogeneous groups of cells. Marker genes were discovered and annotated in previous studies and there are some resources that collect and curate them.
Omnipath is one of the largest available databases of curated prior knowledge. Among its resources, there is PanglaoDB, a database of cell type markers, which can be easily accessed using a wrapper to Omnipath from decoupler.
Note
If you encounter bugs with Omnipath, sometimes is good to just remove its cache using: rm $HOME/.cache/omnipathdb/*
[5]:
# Query Omnipath and get PanglaoDB
markers = dc.get_resource('PanglaoDB')
markers
[5]:
| genesymbol | canonical_marker | cell_type | germ_layer | human | human_sensitivity | human_specificity | mouse | mouse_sensitivity | mouse_specificity | ncbi_tax_id | organ | ubiquitiousness | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | CTRB1 | False | Enterocytes | Endoderm | True | 0.000000 | 0.004394 | True | 0.003311 | 0.020480 | 9606 | GI tract | 0.017 |
| 1 | CTRB1 | True | Acinar cells | Endoderm | True | 1.000000 | 0.000629 | True | 0.957143 | 0.015920 | 9606 | Pancreas | 0.017 |
| 2 | KLK1 | True | Endothelial cells | Mesoderm | True | 0.000000 | 0.008420 | True | 0.000000 | 0.014915 | 9606 | Vasculature | 0.013 |
| 3 | KLK1 | False | Goblet cells | Endoderm | True | 0.588235 | 0.005039 | True | 0.903226 | 0.012408 | 9606 | GI tract | 0.013 |
| 4 | KLK1 | False | Epithelial cells | Mesoderm | True | 0.000000 | 0.008233 | True | 0.225806 | 0.013758 | 9606 | Epithelium | 0.013 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 8456 | SLC14A1 | True | Urothelial cells | Mesoderm | True | 0.000000 | 0.018170 | True | 0.000000 | 0.000000 | 9606 | Urinary bladder | 0.008 |
| 8457 | UPK3A | True | Urothelial cells | Mesoderm | True | 0.000000 | 0.000000 | True | 0.000000 | 0.000000 | 9606 | Urinary bladder | 0.000 |
| 8458 | UPK1A | True | Urothelial cells | Mesoderm | True | 0.000000 | 0.000000 | True | 0.000000 | 0.000000 | 9606 | Urinary bladder | 0.000 |
| 8459 | UPK2 | True | Urothelial cells | Mesoderm | True | 0.000000 | 0.000000 | True | 0.000000 | 0.000000 | 9606 | Urinary bladder | 0.000 |
| 8460 | UPK3B | True | Urothelial cells | Mesoderm | True | 0.000000 | 0.000000 | True | 0.000000 | 0.000000 | 9606 | Urinary bladder | 0.000 |
8461 rows × 13 columns
Since our data-set is from human cells, and we want best quality of the markers, we can filter by canonical_marker and human:
[6]:
# Filter by canonical_marker and human
markers = markers[markers['human'] & markers['canonical_marker'] & (markers['human_sensitivity'] > 0.5)]
# Remove duplicated entries
markers = markers[~markers.duplicated(['cell_type', 'genesymbol'])]
markers
[6]:
| genesymbol | canonical_marker | cell_type | germ_layer | human | human_sensitivity | human_specificity | mouse | mouse_sensitivity | mouse_specificity | ncbi_tax_id | organ | ubiquitiousness | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CTRB1 | True | Acinar cells | Endoderm | True | 1.000000 | 0.000629 | True | 0.957143 | 0.015920 | 9606 | Pancreas | 0.017 |
| 6 | KLK1 | True | Acinar cells | Endoderm | True | 0.833333 | 0.005031 | True | 0.314286 | 0.012826 | 9606 | Pancreas | 0.013 |
| 14 | PRSS3 | True | Acinar cells | Endoderm | True | 0.833333 | 0.028931 | True | 0.028571 | 0.000000 | 9606 | Pancreas | 0.006 |
| 15 | CELA3A | True | Acinar cells | Endoderm | True | 0.833333 | 0.000000 | True | 0.128571 | 0.000000 | 9606 | Pancreas | 0.001 |
| 17 | PRSS1 | True | Acinar cells | Endoderm | True | 1.000000 | 0.005975 | True | 0.028571 | 0.000000 | 9606 | Pancreas | 0.002 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 8260 | TRBC2 | True | T cells | Mesoderm | True | 0.940711 | 0.083362 | True | 0.000000 | 0.000000 | 9606 | Immune system | 0.066 |
| 8266 | TRAC | True | T cytotoxic cells | Mesoderm | True | 1.000000 | 0.131348 | True | 0.000000 | 0.000000 | 9606 | Immune system | 0.042 |
| 8267 | TRAC | True | T cells | Mesoderm | True | 0.972332 | 0.059544 | True | 0.000000 | 0.000000 | 9606 | Immune system | 0.042 |
| 8297 | LCK | True | T cells | Mesoderm | True | 0.648221 | 0.033004 | True | 0.705607 | 0.030902 | 9606 | Immune system | 0.048 |
| 8303 | JUNB | True | T cells | Mesoderm | True | 0.675889 | 0.515822 | True | 0.691589 | 0.356450 | 9606 | Immune system | 0.387 |
691 rows × 13 columns
For this example we will use these markers, but any collection of genes could be used. To see the list of available resources inside Omnipath, run dc.show_resources()
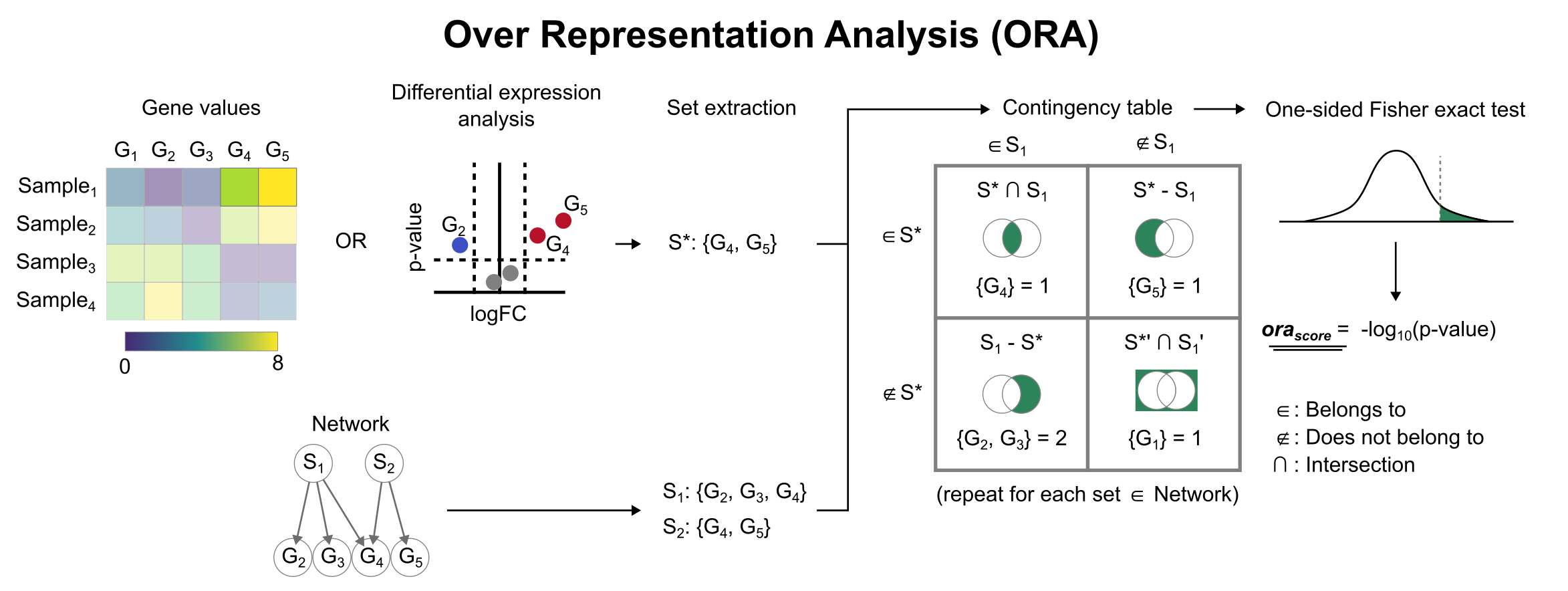
Enrichment with Over Representation Analysis (ORA)
To infer functional enrichment scores we will run the Over Representation Analysis (ora) method. As input data it accepts an expression matrix (decoupler.run_ora) or the results of differential expression analysis (decoupler.get_ora_df). For the former, by default the top 5% of expressed genes by sample are selected as the set of interest (S), and for the latter a user-defined significance filtering can be used. Once we have S, it builds a contingency table using set operations
for each set stored in the gene set resource being used (net). Using the contingency table, ora performs a one-sided Fisher exact test to test for significance of overlap between sets. The final score is obtained by log-transforming the obtained p-values, meaning that higher values are more significant.
We can run ora with a simple one-liner:
[7]:
dc.run_ora(
mat=adata,
net=markers,
source='cell_type',
target='genesymbol',
min_n=3,
verbose=True,
use_raw=False
)
1 features of mat are empty, they will be removed.
Running ora on mat with 2638 samples and 13713 targets for 36 sources.
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 2638/2638 [00:01<00:00, 1411.45it/s]
The obtained scores (-log10(p-value))(ora_estimate) and p-values (ora_pvals) are stored in the .obsm key:
[8]:
adata.obsm['ora_estimate']
[8]:
| source | Acinar cells | Adipocytes | Alpha cells | B cells | B cells naive | Dendritic cells | Ductal cells | Endothelial cells | Enterocytes | Ependymal cells | ... | Pancreatic stellate cells | Pericytes | Plasma cells | Plasmacytoid dendritic cells | Platelets | Podocytes | Proximal tubule cells | Pulmonary alveolar type II cells | Smooth muscle cells | T cells |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAACATACAACCAC-1 | -0.000000 | 0.530653 | 0.885021 | 4.065376 | 2.641014 | 5.720316 | -0.000000 | -0.000000 | -0.000000 | -0.0 | ... | -0.0 | 0.885021 | 2.538999 | 0.437907 | 1.871866 | -0.000000 | -0.000000 | -0.0 | -0.0 | 11.728494 |
| AAACATTGAGCTAC-1 | -0.000000 | -0.000000 | 0.885021 | 5.618952 | 13.894150 | 13.010816 | 2.171878 | 0.827013 | 0.795482 | -0.0 | ... | -0.0 | 0.885021 | 2.538999 | 0.437907 | 2.858756 | -0.000000 | -0.000000 | -0.0 | -0.0 | 2.981581 |
| AAACATTGATCAGC-1 | -0.000000 | 0.530653 | 0.885021 | 1.542530 | 6.107693 | 5.720316 | -0.000000 | 0.298770 | -0.000000 | -0.0 | ... | -0.0 | 0.885021 | 0.569257 | -0.000000 | 5.213387 | -0.000000 | 0.613213 | -0.0 | -0.0 | 9.970169 |
| AAACCGTGCTTCCG-1 | 0.613213 | 1.356127 | 0.885021 | 2.703919 | 2.641014 | 20.564684 | 0.885021 | 0.827013 | -0.000000 | -0.0 | ... | -0.0 | -0.000000 | 1.443226 | 0.437907 | 8.019237 | 0.613213 | -0.000000 | -0.0 | -0.0 | 1.957728 |
| AAACCGTGTATGCG-1 | -0.000000 | -0.000000 | 0.885021 | 1.542530 | 0.349060 | 9.831072 | -0.000000 | 0.298770 | 0.795482 | -0.0 | ... | -0.0 | -0.000000 | 0.569257 | 0.437907 | 2.858756 | -0.000000 | 0.613213 | -0.0 | -0.0 | 1.957728 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| TTTCGAACTCTCAT-1 | 0.613213 | 1.356127 | 0.885021 | 4.065376 | 2.641014 | 22.778754 | 2.171878 | 1.527899 | -0.000000 | -0.0 | ... | -0.0 | 0.885021 | 2.538999 | 1.146495 | 3.976687 | -0.000000 | -0.000000 | -0.0 | -0.0 | 2.981581 |
| TTTCTACTGAGGCA-1 | -0.000000 | -0.000000 | 0.885021 | 5.618952 | 2.641014 | 8.373145 | 0.885021 | 0.298770 | -0.000000 | -0.0 | ... | -0.0 | 0.885021 | 8.735716 | 1.146495 | 3.976687 | -0.000000 | -0.000000 | -0.0 | -0.0 | 1.957728 |
| TTTCTACTTCCTCG-1 | -0.000000 | -0.000000 | 0.885021 | 9.375833 | 15.767762 | 14.738433 | -0.000000 | -0.000000 | 0.795482 | -0.0 | ... | -0.0 | -0.000000 | 3.822062 | 1.146495 | 2.858756 | -0.000000 | 0.613213 | -0.0 | -0.0 | 2.981581 |
| TTTGCATGAGAGGC-1 | -0.000000 | -0.000000 | -0.000000 | 7.375963 | 7.468516 | 9.831072 | -0.000000 | -0.000000 | 0.795482 | -0.0 | ... | -0.0 | -0.000000 | 1.443226 | 0.437907 | 2.858756 | -0.000000 | 1.542530 | -0.0 | -0.0 | 1.957728 |
| TTTGCATGCCTCAC-1 | -0.000000 | -0.000000 | -0.000000 | 1.542530 | 3.687483 | 8.373145 | 0.885021 | 0.827013 | -0.000000 | -0.0 | ... | -0.0 | -0.000000 | -0.000000 | 1.146495 | 2.858756 | -0.000000 | -0.000000 | -0.0 | -0.0 | 11.728494 |
2638 rows × 36 columns
Visualization
To visualize the obtained scores, we can re-use many of scanpy’s plotting functions. First though, we need to extract them from the adata object.
[9]:
acts = dc.get_acts(adata, obsm_key='ora_estimate')
# We need to remove inf and set them to the maximum value observed for pvals=0
acts_v = acts.X.ravel()
max_e = np.nanmax(acts_v[np.isfinite(acts_v)])
acts.X[~np.isfinite(acts.X)] = max_e
acts
[9]:
AnnData object with n_obs × n_vars = 2638 × 36
obs: 'n_genes', 'n_genes_by_counts', 'total_counts', 'total_counts_mt', 'pct_counts_mt', 'leiden'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'leiden', 'leiden_colors'
obsm: 'X_pca', 'X_umap', 'ora_estimate', 'ora_pvals'
dc.get_acts returns a new AnnData object which holds the obtained activities in its .X attribute, allowing us to re-use many scanpy functions, for example:
[10]:
sc.pl.umap(acts, color=['NK cells', 'leiden'], cmap='RdBu_r')
sc.pl.violin(acts, keys=['NK cells'], groupby='leiden')
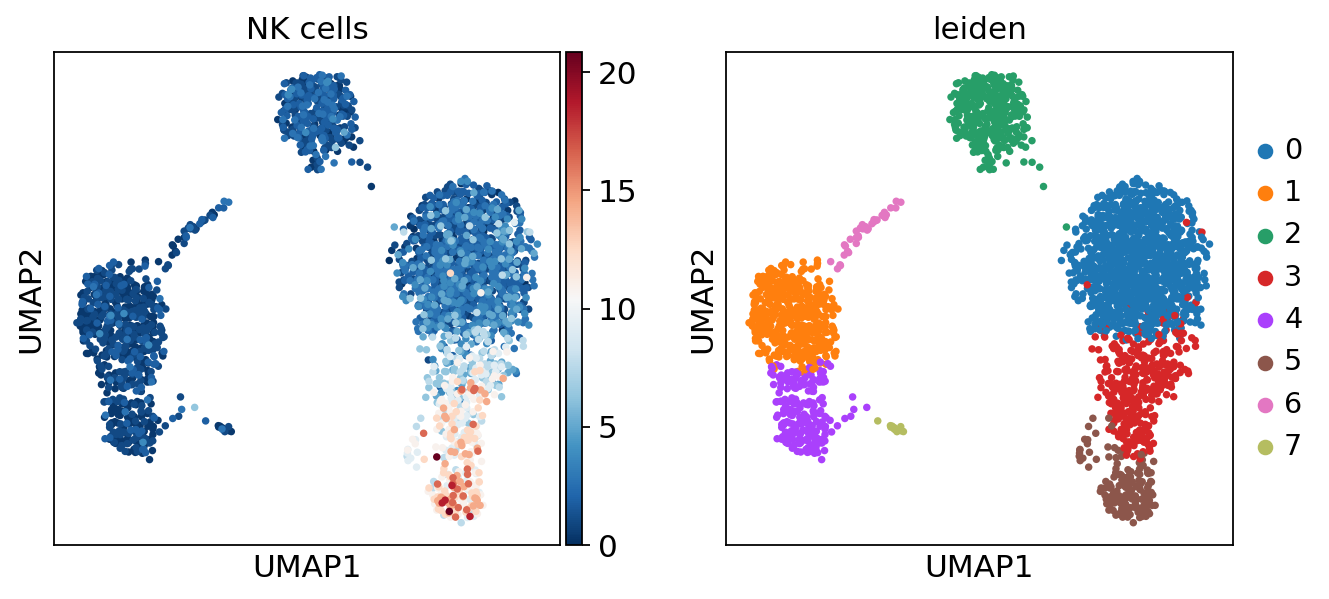
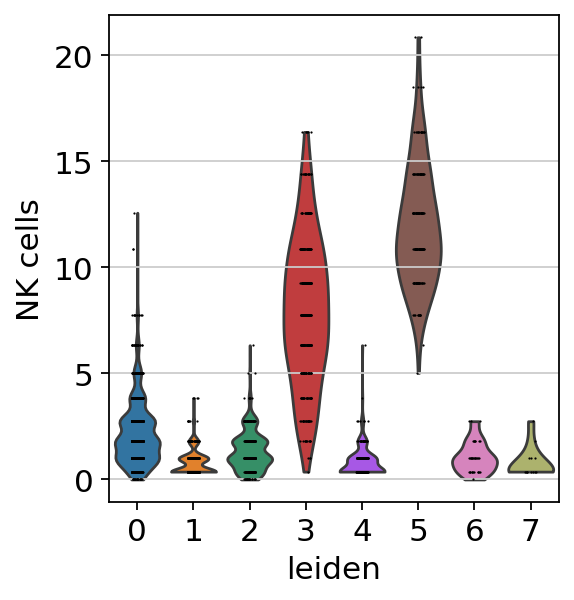
The cells highlighted seem to be enriched by NK cell marker genes.
Annotation
With decoupler we can also identify which are the top predicted cell types per cluster using the function dc.rank_sources_groups. Here, it identifies “marker” cell types per cluster using same statistical tests available in scanpy’s scanpy.tl.rank_genes_groups.
[11]:
df = dc.rank_sources_groups(acts, groupby='leiden', reference='rest', method='t-test_overestim_var')
df
[11]:
| group | reference | names | statistic | meanchange | pvals | pvals_adj | |
|---|---|---|---|---|---|---|---|
| 0 | 0 | rest | T cells | 41.960633 | 5.500564 | 3.985165e-284 | 1.434660e-282 |
| 1 | 0 | rest | Erythroid-like and erythroid precursor cells | 8.247053 | 0.149414 | 2.741401e-16 | 5.805319e-16 |
| 2 | 0 | rest | Pericytes | 6.672327 | 0.121842 | 3.160429e-11 | 5.417879e-11 |
| 3 | 0 | rest | Neurons | 6.057289 | 0.098618 | 1.622875e-09 | 2.655614e-09 |
| 4 | 0 | rest | Oligodendrocytes | 4.901490 | 0.060213 | 1.023526e-06 | 1.535290e-06 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 283 | 7 | rest | Dendritic cells | -2.597187 | -4.715218 | 1.639940e-02 | 4.541372e-02 |
| 284 | 7 | rest | Pericytes | -2.656430 | -0.430002 | 1.390428e-02 | 4.171283e-02 |
| 285 | 7 | rest | B cells naive | -2.846163 | -2.872803 | 1.328863e-02 | 4.171283e-02 |
| 286 | 7 | rest | Ductal cells | -3.244715 | -0.672322 | 5.497033e-03 | 1.978932e-02 |
| 287 | 7 | rest | T cells | -5.148899 | -6.023460 | 1.882849e-04 | 1.129709e-03 |
288 rows × 7 columns
We can then extract the top 3 predicted cell types per cluster:
[12]:
n_ctypes = 3
ctypes_dict = df.groupby('group').head(n_ctypes).groupby('group')['names'].apply(lambda x: list(x)).to_dict()
ctypes_dict
[12]:
{'0': ['T cells', 'Erythroid-like and erythroid precursor cells', 'Pericytes'],
'1': ['Neutrophils', 'Acinar cells', 'Dendritic cells'],
'2': ['B cells naive', 'B cells', 'Plasma cells'],
'3': ['NK cells', 'Gamma delta T cells', 'T cells'],
'4': ['Macrophages', 'Dendritic cells', 'Monocytes'],
'5': ['Gamma delta T cells', 'NK cells', 'Hepatic stellate cells'],
'6': ['Dendritic cells', 'Acinar cells', 'Ductal cells'],
'7': ['Pancreatic stellate cells', 'Platelets', 'Endothelial cells']}
We can visualize the obtained top predicted cell types:
[13]:
sc.pl.matrixplot(acts, ctypes_dict, 'leiden', dendrogram=True, standard_scale='var',
colorbar_title='Z-scaled scores', cmap='RdBu_r')
WARNING: dendrogram data not found (using key=dendrogram_leiden). Running `sc.tl.dendrogram` with default parameters. For fine tuning it is recommended to run `sc.tl.dendrogram` independently.
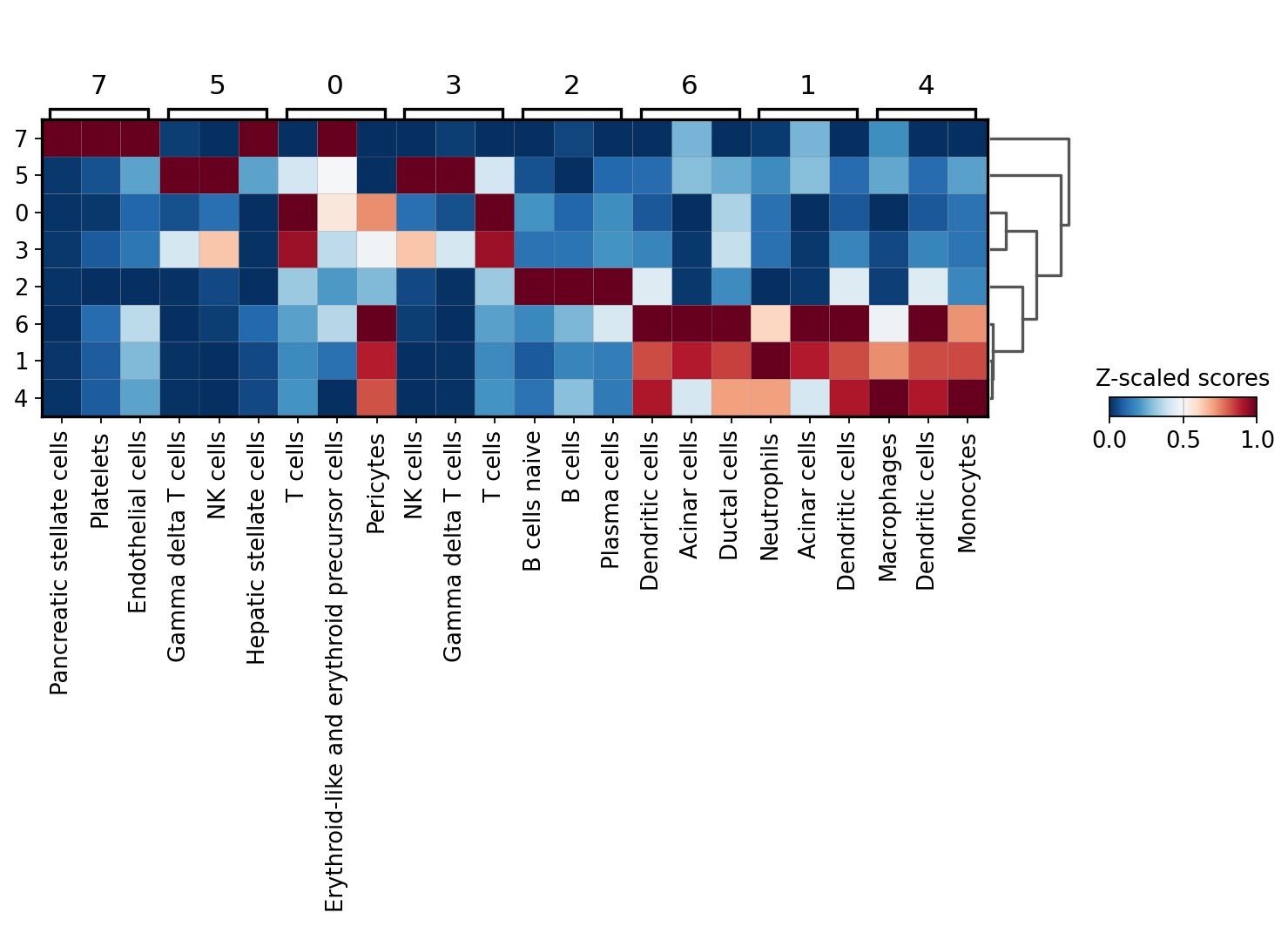
From this plot we see that cluster 7 belongs to Platelets, cluster 4 appear to be NK cells, custers 0 and 3 might be T-cells, cluster 2 should be some sort of B cells and that clusters 6,5 and 1 belong to the myeloid lineage.
We can check individual cell types by plotting their distributions:
[14]:
sc.pl.violin(acts, keys=['T cells', 'B cells', 'Platelets', 'Monocytes', 'NK cells'], groupby='leiden')
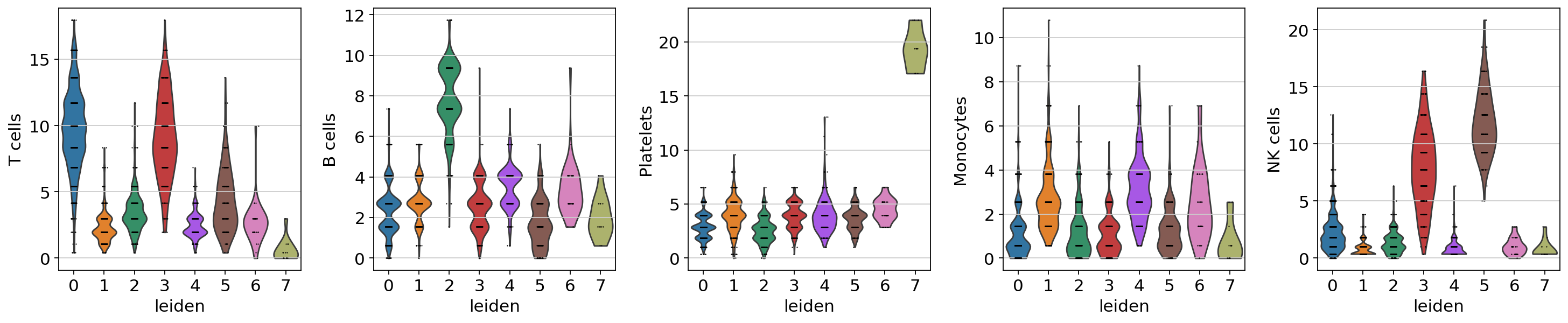
The final annotation should be done manually based on the assessment of the enrichment results. However, an automatic prediction can be made by assigning the top predicted cell type per cluster. This approach does not require expertise in the tissue being studied but can be prone to errors. Nonetheless it can be useful to generate a first draft, let’s try it:
[15]:
annotation_dict = df.groupby('group').head(1).set_index('group')['names'].to_dict()
annotation_dict
[15]:
{'0': 'T cells',
'1': 'Neutrophils',
'2': 'B cells naive',
'3': 'NK cells',
'4': 'Macrophages',
'5': 'Gamma delta T cells',
'6': 'Dendritic cells',
'7': 'Pancreatic stellate cells'}
Once we have selected the top cell type we can finally annotate:
[16]:
# Add cell type column based on annotation
adata.obs['cell_type'] = [annotation_dict[clust] for clust in adata.obs['leiden']]
# Visualize
sc.pl.umap(adata, color='cell_type')
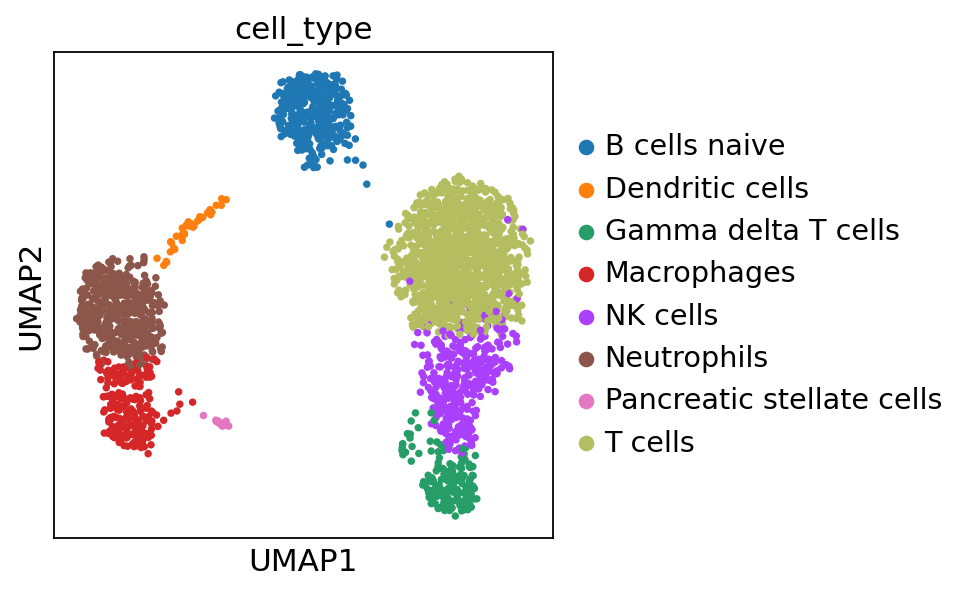
Compared to the annotation obtained by the scanpy tutorial, it is very similar but there are some errors, highlything the limitation of automatic annotation.

Note
Cell annotation should always be revised by an expert in the tissue of interest, this notebook only shows how to generate a first draft of it.